L’Acheiropodie est une maladie génétique rare et complexe, dont la principale caractéristique est l’absence congénitale des mains et des pieds. Cette condition, qui relève d’une anomalie congénitale, suscite un vif intérêt médical en raison des mécanismes précis du développement embryonnaire qu’elle perturbe. Pour comprendre ses particularités, il convient de se pencher sur plusieurs points essentiels :
- Les symptômes cliniques atypiques qui se manifestent dès la naissance.
- Les causes génétiques derrière cette malformation des mains et des pieds.
- Le mode de transmission héréditaire lié à cette pathologie congénitale.
- Les méthodes diagnostiques permettant une identification précoce et sûre.
Cette exploration permettra de mieux appréhender l’impact de cette mutation génétique spécifique, ainsi que les enjeux cliniques pour les patients et leurs familles.
A lire en complément : Échec d'une chirurgie maxillo-faciale : comprendre les causes, les impacts et les solutions possibles
Sommaire
- 1 Symptômes et manifestations cliniques de l’acheiropodie : caractéristique d’une absence de membres
- 2 Origine génétique de l’acheiropodie : mutation du gène LMBR1
- 3 Transmission héréditaire et implications pour les familles : comprendre le trouble héréditaire
- 4 Diagnostic de l’acheiropodie : outils et démarches pour un dépistage sûr
Symptômes et manifestations cliniques de l’acheiropodie : caractéristique d’une absence de membres
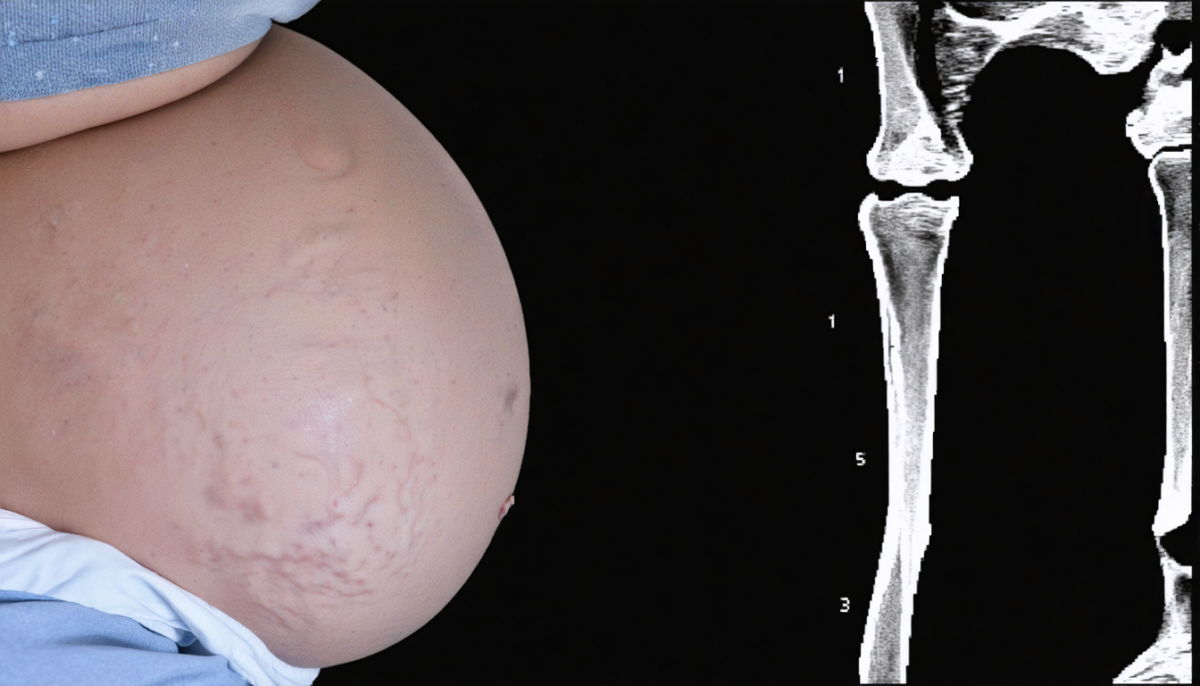
L’acheiropodie, aussi appelée syndrome tétra-amélie distale, se traduit par une absence totale des extrémités des membres, remplacées par des moignons arrondis sans formation des mains ni des pieds. Cette anomalie apparaît tôt dans le développement embryonnaire, entre la 4ᵉ et la 8ᵉ semaine de grossesse, période cruciale pour la morphogenèse des membres.
Voici quelques caractéristiques observées chez les personnes atteintes :
A découvrir également : Les mensonges à la médecine du travail : quels dangers et impacts pour votre santé professionnelle ?
- Moignons arrondis: les bras et jambes s’arrêtent brusquement avant les structures distales, avec une peau souvent cicatricielle ou hyperpigmentée.
- Préservation de la motricité proximale: les épaules et hanches conservent une mobilité normale, permettant certains mouvements fonctionnels.
- Compromission fonctionnelle: l’absence des mains et des pieds entraîne des difficultés majeures dans la préhension, la manipulation et la locomotion.
- Absence d’atteinte neurologique: les capacités cognitives restent intactes, ce qui est rassurant pour l’accompagnement éducatif.
Les registres médicaux comptabilisent moins d’une centaine de cas documentés mondialement, soulignant la rareté extrême de cette maladie.
Origine génétique de l’acheiropodie : mutation du gène LMBR1
La maladie découle d’une mutation génétique spécifique affectant le gène LMBR1, situé sur le chromosome 17. Ce gène est un acteur majeur dans la régulation des étapes fondamentales du développement embryonnaire des membres, notamment la formation des structures distales comme les mains et les pieds.
Plusieurs types de mutations ont été identifiés :
- Mutations ponctuelles : modifications mineures mais impactant la fonction normale du gène.
- Délétions : pertes de segments du gène empêchant la production de protéines fonctionnelles.
- Insertions : ajout de séquences anormales perturbant les cascades de signalisation.
Ces altérations génétiques bloquent la morphogenèse des bourgeons des membres, ce qui explique l’absence des extrémités chez les nouveau-nés affectés.
Transmission héréditaire et implications pour les familles : comprendre le trouble héréditaire
Le mode de transmission de l’acheiropodie est autosomique récessif. Pour qu’un enfant développe la maladie, il doit hériter de deux copies mutées du gène LMBR1 – une venant de chaque parent. Les parents, s’ils possèdent une seule copie mutée, sont porteurs sains sans symptômes apparents.
Ce schéma génétique conduit aux probabilités suivantes lors d’une grossesse :
| Statut de l’enfant | Probabilité |
|---|---|
| Enfant atteint | 25 % |
| Enfant porteur sain | 50 % |
| Enfant non porteur | 25 % |
Un conseil génétique est vivement conseillé, notamment pour les couples avec antécédents familiaux ou issus de communautés où la consanguinité est plus fréquente. Ce conseil facilite la prise de décisions éclairées autour de la reproduction.
Diagnostic de l’acheiropodie : outils et démarches pour un dépistage sûr
Le diagnostic peut être posé à deux moments clés :
- Diagnostic prénatal : grâce à l’échographie de haute résolution, l’absence des mains et pieds est parfois visible dès la 18ᵉ semaine de grossesse. Cette observation est essentielle pour anticiper la prise en charge à la naissance.
- Examen postnatal : l’inspection clinique détecte immédiatement la malformation. Des examens radiographiques confirment l’absence des structures osseuses distales, tandis qu’un test génétique permet d’identifier la mutation du gène LMBR1.
Afin de réduire les risques de transmission dans les familles affectées, des techniques comme le diagnostic préimplantatoire (DPI) sont disponibles, offrant la possibilité de choisir des embryons sans mutation lors d’une fécondation in vitro.
Cette démarche, accompagnée par un suivi médical et un accompagnement psychologique, transforme l’approche autour de cette pathologie congénitale en offrant des perspectives d’espoir et d’adaptation.